【SEACells Tutorial(一):基于SEACells的scATAC-seq经典下游分析】_seacells infers transcriptional and epigenomic cel-程序员宅基地
SEACells Tutorial(一):基于SEACells的scATAC-seq经典下游分析
写在前面:
目前,单细胞基因组学数据的细胞分辨率与大多数分析的集群级分辨率之间存在着根本的脱节,简单来说就是在聚类分析的过程中,通常会导致集群级分辨率,这意味着一组细胞被归为同一类别,而忽略了它们内部的细微差异。
为了克服数据的稀疏性和噪声的问题,我们通常会把metacells作为一个有效的解决办法。通俗来说,metacells是单细胞测序数据的细胞分组,即在不同深度下对单细胞进行聚合,其细胞内的变异都是由技术因素引起的,而非生物学因素,这样就使得细胞的均匀性大大提高。
2023年3月27日,在nature biotechnology上发布了一边文章,作者给出了一种计算metacells的算法,可从单细胞基因组学数据中推断出转录和表观基因组的细胞状态。在这里我们对作者发布的SEACells进行复现,期望能够在实践中加以应用。
备注:本教程代码部分在Google Colab运行环境中完成,Google Colab是Google提供的一个Jupyter Notebook式的交互环境。
文章目录
概述
本文章主要为利用SEAcells进行scATAC-seq的下游分析提供一个教程,包含了四部分的内容:gene-peak associations的计算、highly regulated genes的识别、ATAC gene 得分计算以及基因可及性得分(gene accessibility scores),这大概包含了ATAC分析下游的大部分分析流程。
一、准备工作
#载入包
import numpy as np
import pandas as pd
import scanpy as sc
import SEACells
import matplotlib
import matplotlib.pyplot as plt
import seaborn as sns
#画图设置
%matplotlib inline
sns.set_style('ticks')
matplotlib.rcParams['figure.figsize'] = [4, 4]
matplotlib.rcParams['figure.dpi'] = 100
二、加载数据
我们使用 scanpy Anndata 对象作为加载和过滤数据的首选模式。
# !mkdir data/
# !wget https://dp-lab-data-public.s3.amazonaws.com/SEACells-multiome/cd34_multiome_rna.h5ad -O data/cd34_multiome_rna.h5ad # RNA data
# !wget https://dp-lab-data-public.s3.amazonaws.com/SEACells-multiome/cd34_multiome_atac.h5ad -O data/cd34_multiome_atac.h5ad # ATAC data
该数据集包含 RNA 和 ATAC 模式作为两个不同的 Anndata 对象。ATAC 数据集包含预先计算的 SEACell 元细胞
(如何计算seacell后续SEACells系列文章会提到)
数据集介绍
这是过滤后的由 CD34+ 分选出来的的骨髓细胞数据集,未进行标准化,用于分析人类造血功能,详细信息可根据上述下载链接进行查看。
# 用scanpy加载数据集
rna_ad = sc.read('data/cd34_multiome_rna.h5ad')
atac_ad = sc.read('data/cd34_multiome_atac.h5ad')
# ATAC的参考细胞类型
sc.pl.scatter(atac_ad, basis='umap', color='celltype', frameon=False)

# SEACells的metacells计算结果
SEACells.plot.plot_2D(atac_ad, key='X_umap', colour_metacells=False)

三、提取元细胞
ATAC 和 RNA 单细胞 Anndata 对象应包含同一组细胞。仅相同细胞将用于下游分析。
atac_meta_ad, rna_meta_ad = SEACells.genescores.prepare_multiome_anndata(atac_ad, rna_ad, SEACells_label='SEACell')
atac_meta_ad,rna_meta_ad
AnnData object with n_obs × n_vars = 99 × 246113
obs: 'n_counts'
var: 'GC_bin', 'counts_bin', 'n_cells'
uns: 'log1p'
obsm: 'X_svd'
layers: 'raw'
AnnData object with n_obs × n_vars = 99 × 12464
obs: 'n_counts'
uns: 'log1p'
layers: 'raw'
四、Gene-peak correlations计算
要计算基因表达和基因附近峰的可及性的相关性,需要:
1、带有基因注释的 GTF 文件及基因周围的基因组跨度信息以测试相关性
2、染色体名称应编号为 1:、2:形式
#基因组文件获取
! wget https://dp-lab-data-public.s3.amazonaws.com/SEACells-multiome/hg38.gtf -O data/hg38.gtf
# 在此示例中,我们计算自此以来的前十个基因的基因峰值相关性,方便进行演示
# 这个过程是计算密集型的
gene_set = rna_meta_ad.var_names[:10]
gene_peak_cors = SEACells.genescores.get_gene_peak_correlations(atac_meta_ad, rna_meta_ad,
path_to_gtf='data/hg38.gtf',
span=100000,
n_jobs=1,
gene_set=gene_set)
#查看运行结果
gene_peak_cors['FAM41C'].head()

五、高度调控基因的计算
对于下游分析,完整的基因峰值相关性结果可作为 pickle 文件提供,网址为 https://dp-lab-data-public.s3.amazonaws.com/SEACells-multiome/cd34_multiome_gene_peak_cors.p
#基因组文件获取
! wget https://dp-lab-data-public.s3.amazonaws.com/SEACells-multiome/cd34_multiome_gene_peak_cors.p -O data/cd34_multiome_gene_peak_cors.p
gene_peak_cors = pd.read_pickle('data/cd34_multiome_gene_peak_cors.p')
# 高度调控的基因,即与多个峰相关的基因,可以使用 get_gene_peak_assocations 函数来识别。get_gene_peak_assocations 返回与每个基因相关的显着峰的数量
peak_counts = SEACells.genescores.get_gene_peak_assocations(gene_peak_cors,
pval_cutoff=1e-1,
cor_cutoff=0.1)
参数说明:
pval_cutoff=1e-1: 这个参数设置了一个p值截止值。相关性的p值大于这个截止值(在这种情况下是0.1)的基因峰值相关性可能被过滤掉。这意味着只有p值小于或等于0.1的相关性被认为是显著的。
cor_cutoff=0.1: 这个参数设置了一个相关系数截止值。相关性系数小于这个截止值(在这种情况下是0.1)的基因峰值相关性可能被过滤掉。这意味着只有相关系数大于或等于0.1的相关性被认为是显著的。
#绘制分布图以识别具有较高调控程度的基因
plt.scatter(np.arange(len(peak_counts)),
np.sort(peak_counts), s=20)
sns.despine()
plt.xlabel('Gene rank')
plt.ylabel('No. of correlated peaks')

六、基因得分
基因分数计算为相关峰的可及性的加权和,并且可以使用 get_gene_scores 计算。
gene_scores = SEACells.genescores.get_gene_scores(atac_meta_ad,
gene_peak_cors,
pval_cutoff=1e-1,
cor_cutoff=0.1)
gene_scores 是一个 pandas.Data Frame,其中元细胞为行,基因为列。
这可用于任何下游分析,例如聚类、可视化等。
七、Gene-accessibility计算
元细胞中的开放峰识别
基因分数计算为相关峰的可及性的加权和,并且可以使用 get_gene_scores 计算。
该函数还需要低维嵌入,例如 X_svd、X_umap等等。
此处我们应用scATAC 的 SVD 来进行下面的分析
# 用原始计数矩阵创建一个元细胞的anndata数据
atac_meta_ad = SEACells.core.summarize_by_SEACell(atac_ad, SEACells_label='SEACell')
atac_meta_ad.obs['n_counts'] = np.ravel(atac_meta_ad.X.sum(axis=1))
# 加入svd低维嵌入
seacell_label = 'SEACell'
sc_svd = pd.DataFrame(atac_ad.obsm['X_svd'], index=atac_ad.obs_names)
atac_meta_ad.obsm['X_svd'] = sc_svd.groupby(atac_ad.obs[seacell_label]).mean().loc[atac_meta_ad.obs_names, :]
# 确定每个元细胞中的开放峰
SEACells.accessibility.determine_metacell_open_peaks(atac_meta_ad, peak_set=None, low_dim_embedding='X_svd', pval_cutoff=1e-2,
read_len=147, n_neighbors=3, n_jobs=1)
# 该函数会将“Open Peaks”添加到 Anndata 层,并且是一个二进制矩阵,指示元细胞中峰是开放的还是封闭的
基因可及性指标
开放峰用于计算基因可及性度量,该度量表示相关开放峰的分数。
注意:只有当每个基因都有足够数量的开放峰时,此指标才是可靠的。
建议仅用于高度调控的基因,即我们第五部分计算得到的结果。
# 根据肘点选择基因,此处的参考图为高度调控基因的分布图
high_reg_genes = peak_counts.index[peak_counts > 9]
# 计算基因可及性
SEACells.accessibility.get_gene_accessibility(atac_meta_ad, gene_peak_cors,
gene_set=high_reg_genes, pval_cutoff=1e-1, cor_cutoff=0.1)
# 此函数会将“基因可访问性”添加到 Anndata `.obsm` 字段
可视化
# 首先加入umap以可视化基因可及性
rna_umap = pd.DataFrame(rna_ad.obsm['X_umap'], index=rna_ad.obs_names)
rna_meta_ad.obsm['X_umap'] = rna_umap.groupby(atac_ad.obs[seacell_label]).mean().loc[rna_meta_ad.obs_names, :].values
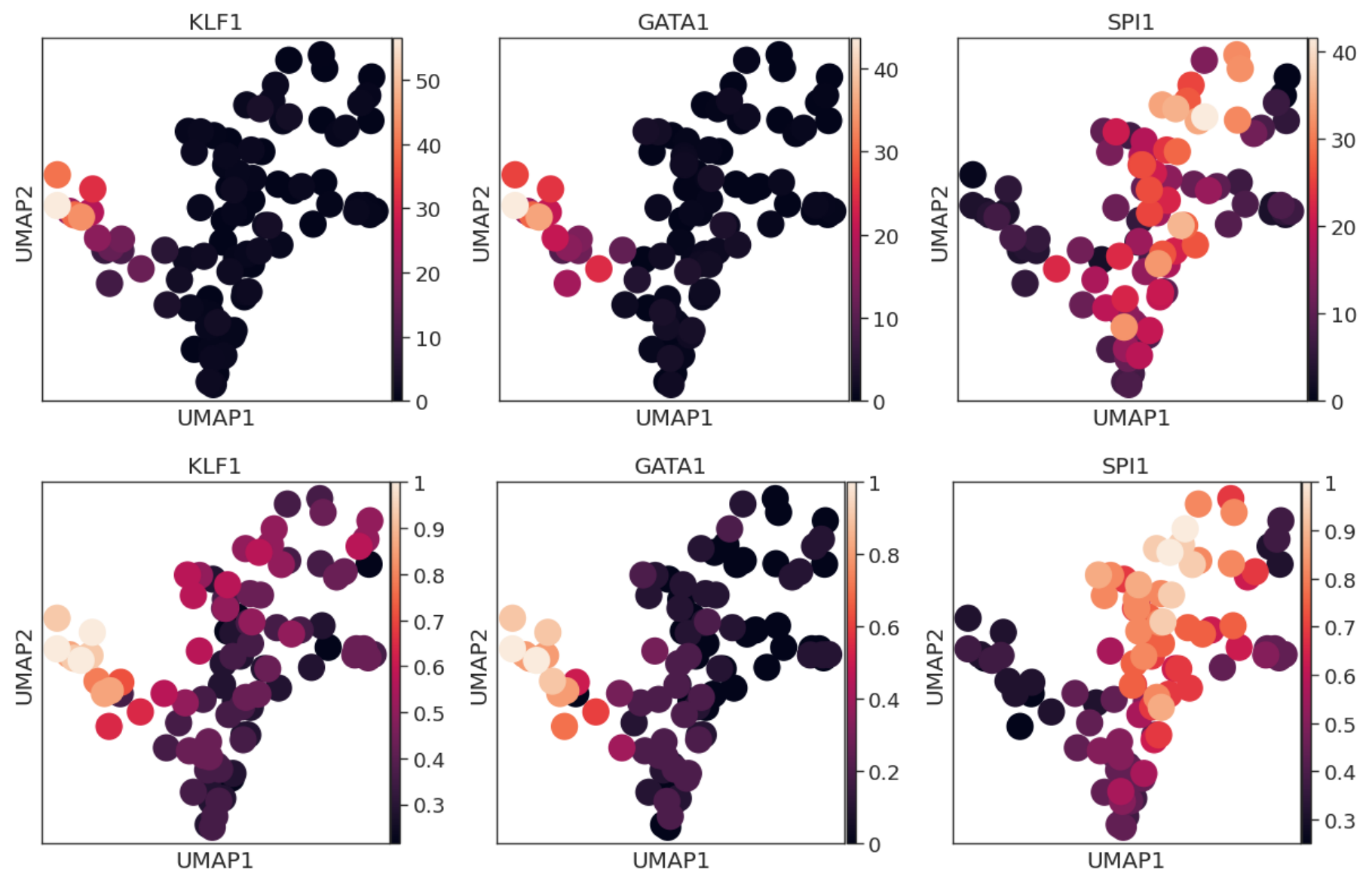
genes = ['KLF1', 'GATA1', 'SPI1']
# Copy accessibility to RNA meta anndata
temp = rna_meta_ad[:, genes]
temp.layers['GeneAccessibility'] = atac_meta_ad[rna_meta_ad.obs_names].obsm['GeneAccessibility'][genes].values
# Plot expression
sc.pl.scatter(rna_meta_ad, basis='umap', color=genes)
# Plot accessibility
sc.pl.scatter(temp, basis='umap', color=genes, layers='GeneAccessibility')
Reference
[1]https://github.com/dpeerlab/SEACells
[2]Persad, S., Choo, ZN., Dien, C. et al. SEACells infers transcriptional and epigenomic cellular states from single-cell genomics data. Nat Biotechnol (2023).
智能推荐
Learning and Using Jakarta Digester-程序员宅基地
文章浏览阅读601次。今天有空再度学习Struts1.3.9的源码,感觉对org.apache.commons.digester.Digester的认识还很少,上网看了一篇文章讲的比较好,特此转载! 文章出处:http://www.onjava.com/pub/a/onjava/2002/10/23/digester.html?page=1 文章中的案例解释: //生成一个digester。主要
自己封装的文件服务苹果手机无法播放视频-程序员宅基地
文章浏览阅读370次。自己封装的文件服务苹果手机无法播放视频
网鼎杯第四场 shenyue2 writeup_ctf shenyue2-程序员宅基地
文章浏览阅读987次。1. shenyue2题目分析这是一道RSA相关的密码学题目,给出了RSA相关的公钥(n,e)(n,e)(n,e),并且给出了额外的两个参数:一个已知素数rrr以及kkk,并且有如下关系: k=(p−r)dk=(p−r)dk=(p-r)d 其实这是一道2018 CodeGate CTF 的原题,直接按照CTF-WIKI中介绍的解法就可以求出来。而我写这篇文章的目的是向大家介绍另外一种方法,..._ctf shenyue2
将11.x.x升级至16.x.x不成功的一系列问题(二)node-sass sass-loader需安装指定版本_node-sass 升级-程序员宅基地
文章浏览阅读1k次,点赞20次,收藏20次。先根据node版本先锁定node-sass版本 然后再来回切换sass-loader版本 这玩应你就试吧 一试一个一个不吱声_node-sass 升级
python openpyxl ValueError: Value does not match pattern ^[$]?([A-Za-z]{1,3})[$]?(\d+)(:[$]?-程序员宅基地
文章浏览阅读6.7k次。这种问题是因为sheet名称有问题,一般是名字两边有空格?解决:新建一个sheet,然后查看代码,输进去这些,一运行就出来了Sub listSheetName()i = 1For Each sSheet In Application.SheetsCells(i, 1).Value = sSheet.Namei = i + 1NextsSheetEndSub获取到所有..._valueerror: value does not match pattern ^[$]?([a-za-z]{1,3})[$]?(\d+)(:[$]?
Linux 下 对‘pthread_create’未定义的引用_linux 中pthread_create()未定义-程序员宅基地
文章浏览阅读1k次,点赞2次,收藏2次。pthread库不是Linux系统默认的库,连接时需要使用库libpthread.a,在编译中要加-lpthreadgcc xxx.c.pp -lpthread 针对直接编译的target_link_libraries(thread libpthread.so) 针对cmake的这里thread是我的二进制文件..._linux 中pthread_create()未定义
随便推点
【hbase】hbase使用MR统计行数指定yarn队列,及后续遇到的问题_org.apache.hadoop.hbase.mapreduce.rowcounter 增加参数-程序员宅基地
文章浏览阅读1.1k次。一、前言最近需要统计一张hbase表的条数,网上的很多案例都是使用MR的方式来进行统计,所以我们也采用这个方式。但是在实施过程中,遇到一些问题。使用MR去统计时,如果不指定队列,那么就会使用默认的YARN队列,而我们的默认队列是完全没有资源的。网上关于指定YARN队列的文章也比较少,这里整理并记录一下。二、准备这里我们使用 hbase.RowCounter包执行MR的任务。[hbase@bi-hadoop02 ~]$ hbase org.apache.hadoop.hbase.mapreduce_org.apache.hadoop.hbase.mapreduce.rowcounter 增加参数
FAILED: Execution Error, return code 1 from org.apache.hadoop.hive.ql.exec.DDLTask. MetaException(me-程序员宅基地
文章浏览阅读1.8k次。FAILED: Execution Error, return code 1 from org.apache.hadoop.hive.ql.exec.DDLTask. MetaException(message:org.apache.hadoop.hbase.client.RetriesExhaustedException: Can't get the locations at org.apache.hadoop.hbase.client.RpcRetryingCallerWithReadReplicas_failed: execution error, return code 1 from org.apache.hadoop.hive.ql.exec.d
python计算机毕设【附源码】医用仓库管理系统(django+mysql+论文)-程序员宅基地
文章浏览阅读518次,点赞6次,收藏10次。在数据库管理工具的选择上,使用了Navicat 11,这是一个用户友好且功能强大的数据库管理软件,它支持多种数据库系统,包括MySQL,并提供了图形化界面,使得数据库的管理和维护工作更加便捷。开发环境方面,我们选择了PyCharm作为主要的集成开发环境(IDE),它提供了丰富的Python开发工具和插件,支持Django框架,有助于提高开发效率和代码质量。此外,系统还可以记录操作人员的操作记录,便于追踪和审计。提高物资利用率:通过对医用物资的有效管理,可以避免物资的浪费和积压,提高物资利用率。
WIN10 LTSC 2019 安装新版Mircosoft Edge浏览器,解决无法安装问题_ltsc安装不了edge-程序员宅基地
文章浏览阅读1.8w次,点赞3次,收藏7次。WIN10 LSTC 2019 安装新版Mircosoft Edge浏览器,解决无法安装问题自己的笔记本一直使用WIN10 2019 LSTC(MSDN下载的)这一版本,因为这一版本真的太轻快简洁了,最近换电脑也是安装的Win10 LSTC 2019,但是安装完毕后在安装新版edge浏览器的时候提示系统版本低安装不了,自己的旧电脑就是LSTC却安装上,不知道什么原因,后来回想了一下旧电脑的使用过程并对新旧电脑的系统做了详细对比,发现是旧电脑上的系统补丁包版本高,而新电脑是刚安装的系统,补丁包还没有过升级_ltsc安装不了edge
Pluto SDR环境搭建libiio/libad9361-iio/GNU Radio/gr-iio(Ubuntu)_gnuradio 3.8.2 如何支持adalm pluto-sdr windows-程序员宅基地
文章浏览阅读784次,点赞11次,收藏10次。ADI前些年推出的ADALM-PLUTO SDR设备由于其轻便灵活的特点,外加价格相比于专业无线电相当实惠,受到了很多开源社区的欢迎,也诞生了许多的应用,如跟踪GPS、伪造GPS实现硬件级虚拟定位、电子钥匙重发攻击等(这些实际上HackRF做的更多)。同时对于学习通信的师生和对无线电感兴趣的业余玩家,也是个很不错的选择。国内购买纯原版Pluto SDR有些困难,但好在国内也有很多企业或团队基于某些成熟的SDR平台衍生出的性能更强,适用固件更多的软件无线电平台,价格也并非难以承受。_gnuradio 3.8.2 如何支持adalm pluto-sdr windows
SIM卡、USIM卡、UICC卡、eSIM卡的区别_uhimpc-程序员宅基地
文章浏览阅读2.8k次。SIM的英文全称是“Subscriber Identity Module”,即“用户身份模块”。它的主要作用是在移动终端设备与网络通讯时提供身份识别信息及存储数据,大家比较容易理解的就是我们的电话号码(身份识别信息)是与SIM卡直接绑定的,还有SIM卡还可以存储电话号码、短消息等数据。COMPRION公司的测试用SIM卡现在的3G与4G移动系统里,准确地说SIM是一个应用的概念,..._uhimpc