数据挖掘实验-主成分分析与类特征化_主成分分析的目的和作用-程序员宅基地
技术标签: 数据挖掘
数据集&代码![]() https://www.aliyundrive.com/s/Jtcuion5iNC
https://www.aliyundrive.com/s/Jtcuion5iNC
一.主成分分析
1.实验目的
-
了解主成分分析的目的,内容以及流程。
-
掌握主成分分析,能够进行编程实现。
2.实验原理
主成分分析的目的
主成分分析就是把原有的多个指标转化成少数几个代表性较好的综合指标,这少数几个指标能够反映原来指标大部分的信息(85%以上),并且各个指标之间保持独立,避免出现重叠信息。主成分分析主要起着降维和简化数据结构的作用。
主成分分析的数学模型
假设所讨论的实际问题中,有p个指标,把这p个指标看作p个随机变量,记为X1,X2,…,Xp,主成分分析就是要把这p个指标的问题,转变为讨论 m 个新的指标F1,F2,…,Fm(m<p),按照保留主要信息量的原则充分反映原指标的信息,并且相互独立。
![]()
主成分分析的做法是寻求原指标的线性组合Fi。
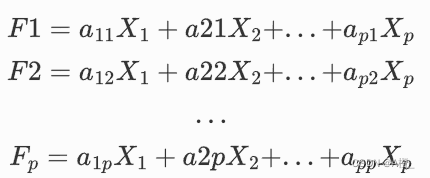
满足如下条件:
-
主成分的系数平方和为1:
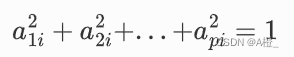
-
主成分之间相互独立,无重叠的信息:
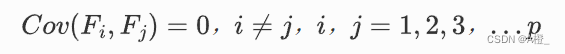
-
主成分之间的方差依次递减:

求解主成分的过程
-
求样本均值\overline{X}和样本协方差矩阵S;
-
求S的特征根\lambda
-
求特征根所对应的单位特征向量
-
写出主成分的表达式
![]()
其中主成分选取时,根据累积贡献率的大小取前面m个主成分。
选取原则:
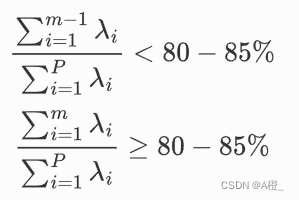
3.实验过程
R语言内置了主成分分析的计算函数,也有其他的包可以进行主成分分析,实验中使用R语言,采用内置的PCA主成分分析函数进行主成分分析。以下以对癌症数据集BLCA的数据进行主成分分析为例,说明完成主成分分析的过程,其他数据集的方法是相同的。
(1)环境准备
安装并导入用于多元统计分析可视化的factoextra包。
options(repos=structure(c(CRAN="https://mirrors.tuna.tsinghua.edu.cn/CRAN/")))
install.packages("factoextra",dependencies = TRUE)
library(factoextra)将数据导入。
data <- read.csv('D:\\Files\\文档\\HNU5\\数据挖掘\\数据集\\BLCA\\rna.csv') 进行转置,将样本放在行,属性(gene_id)放在列上。
data <- t(data)(2)进行主成分分析
使用R语言内置的prcomp函数进行主成分分析:
cancer.pr <- prcomp(data)R语言中的prcomp函数可以有另外两个参数:
-
SCALE:设置为true,则在进行主成分分析之前将数据进行标准化。
-
RANK:主成分个数。
当各变量之间存在不同量纲时,就需要使用标准化。在对本书聚集进行分析时,考虑到数据都是样本的基因表达值,假设量纲是相同的,不进行标准化。
使用summary函数查看主成分贡献率:
summary(cancer.pr)部分结果如下:
其中Standard deviation表示标准差,Proportion of Variance表示单主成分贡献率,Cumulative Proportion表示累积贡献率。根据主成分的选取原则,选取的m个主成分的累积贡献率应该大于80%,因此可以看出,需要选择前116个主成分,此时主成分的累计贡献率为80.15%,可以反映原来指标大部分的信息。

进行主成分选取时,还可以生成碎石图,查看最大到最小排列的贡献值:
fviz_eig(cancer.pr, addlabels = TRUE, ylim = c(0, 100)) 
这里可以看出,最大的主成分贡献值有15%,大部分主成分贡献值都比较小,因此才需要前116个主成分才能使累积贡献率达到80%,这也说明原数据无法降维到很小的维度。
还可以绘制一个变量相关性可视化图,相关图中越远离原点,主成分PC对变量的代表性越高(相关性强),靠近的变量为正相关。
fviz_pca_var(cancer.pr)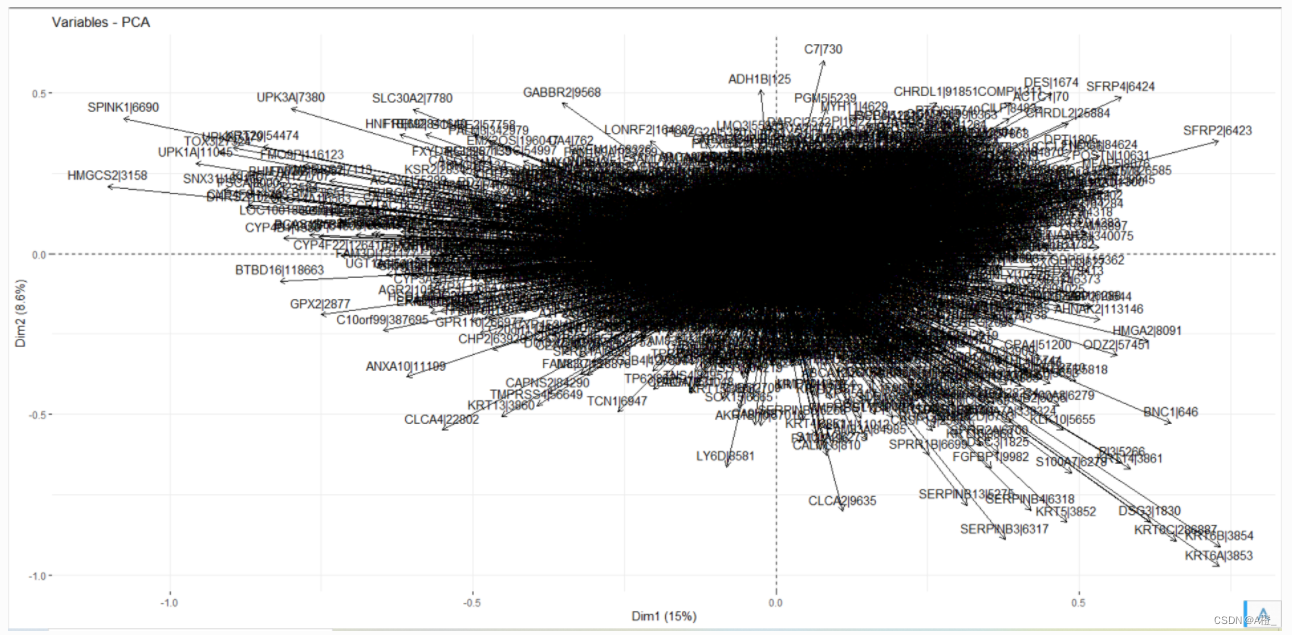
还有一些其他的绘图函数和主成分分析结果的信息,表示主成分对变量的代表性强弱,每个变量对特定主成分的贡献等,这里由于主成分分析后数据维度仍然很高,不做相关的分析。
(4)计算主成分数据
主成分分析主要起着降维和简化数据结构的作用。经过以上对主成分的求解,已经确定可以保留前20个主成分,只需要计算各个样本的主成分值,此后对数据的分析只需要基于主成分数据,不需要使用原始数据了。最后将结果导出,就完成了数据的主成分分析。
cancer.pca <- predict(cancer.pr)[,1:116]
write.table (cancer.pca, file ="D:\\Files\\文档\\HNU5\\数据挖掘\\数据集\\BLCA\\pca.csv", sep =",", row.names =TRUE, col.names =TRUE, quote =TRUE)对于主成分分析后的样本,可以进行坐标可视化:
fviz_pca_ind(cancer.pr)#score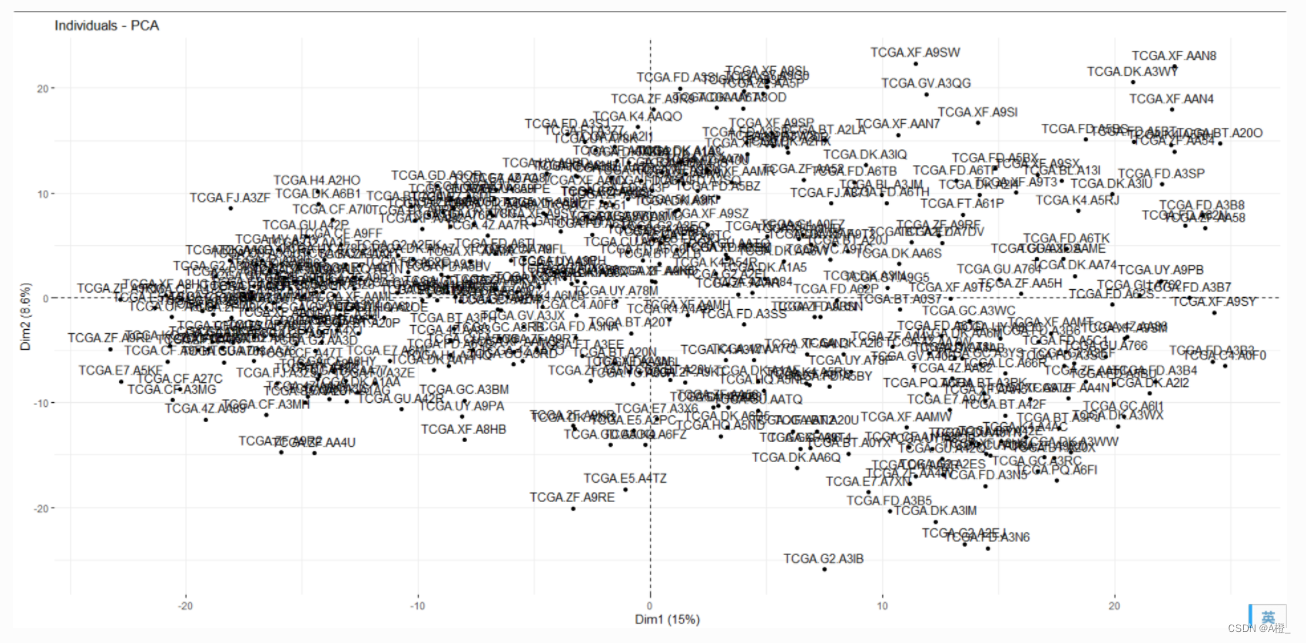
对于不同癌症类型的数据集,进行主成分分析后:
| 类型 | 主成分数 | 累积贡献率 |
|---|---|---|
| BLCA | 116 | 80.15% |
| BRCA | 212 | 80.072% |
| KIRC | 114 | 80.125% |
| LUAD | 140 | 80.099% |
| PAAD | 45 | 80.15% |
4.实验结果分析
对不同的数据集进行主成分分析后,数据维度明显降低了,体现了主成分分析进行数据降维的作用。但仍需保留很多主成分才能满足累积贡献率达到80%的要求,这体现了数据的复杂程度。主成分分析后的各个数据集没有相同的属性,不清楚如何对不同癌症类型的主成分分析后的数据进行比较,因此没有完成。
二.类概念描述及特征化分析
1.实验目的
-
了解类特征化和类对比分析目的,内容以及流程。
-
掌握类特征化和类对比分析,能够编程实现。
-
掌握属性相关分析,实现基于信息增益的属性相关分析。
2.实验原理
类特征化和类对比分析
概念(类)描述是描述性数据挖掘的最基本形式。类描述由类特征和比较组成。
-
类特征化:汇总并描述称作目标类的数据集。
-
类(比较)对比:汇总并将一个称作目标类数据集与称作对比类的其他数据集相区别。
属性相关分析
-
通过识别不相关或者是弱相关的属性,将他们排除在概念描述过程之外,从而确定哪些属性应当包含在类特征化和类比较中。
-
属性相关分析的基本思想是计算某种度量,用于量化属性与给定类或概念的相关性。可采用的度量包括信息增益、不确定性和相关系数。
信息增益
信息增益通过计算一个样本分类的期望信息和属性的熵来获得一个属性的信息增益,判定该属性与当前的特征化任务的相关性。
信息增益的计算方法如下:
S是一个训练样本的集合,该样本中每个集合的类编号已知。每个样本为一个元组。有个属性来判定某个训练样本的类编号。
假设S中有m个类,共S个训练样本,每个类Ci有Si个样本,那么任意一个样本属于类Ci的概率是Si/S,那么用来分类一个给定样本的期望信息是:
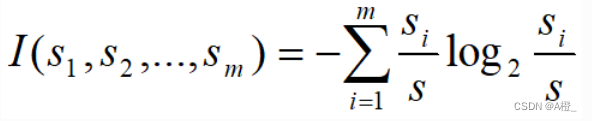
一个有v个值的属性A{a1,a2,...,av}可以将S分成v个子集{S1,S2,...,Sv},其中Sj包含S中属性A上的值为aj的样本。假设Sj包含类Ci的sij个样本。根据A的这种划分的期望信息称为A的熵。
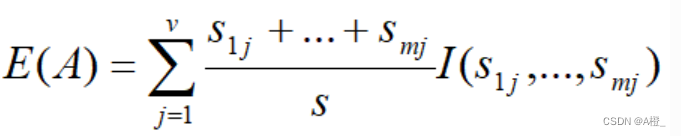
A上该划分的获得的信息增益定义为:

具有高信息增益的属性,是给定集合中具有高区分度的属性。所以可以通过计算S中样本的每个属性的信息增益,来得到一个属性的相关性的排序。就可以识别不相关或者是弱相关的属性,将其排除在概念描述过程外。
包含属性相关分析的类特征化的过程
-
数据收集
-
通过查询处理收集数据库中相关的数据,并将其划分为一个目标类和一个或多个对比类。
-
-
预相关分析
-
识别属性和维的集合
-
对有大量不同值的属性进行删除或概化
-
产生候选关系
-
-
使用选定的相关分析度量删除不相关弱相关的属性
-
使用相关分析度量(信息增益),评估候选关系中的每个属性
-
根据所计算的相关性对属性进行排序
-
低于临界值的不相关和弱相关的属性被删除
-
产生初始目标类工作关系
-
-
产生概念描述
包含属性相关分析的类对比分析的过程
类对比分析的过程如下:
-
数据收集:
通过查询处理收集数据库中相关的数据,并将其划分为一个目标类和一个或多个对比类。
-
维相关分析:
使用属性相关分析方法,使我们的任务中仅包含强相关的维。
-
同步概化
同步的在目标类和对比类上进行概化,得到主目标类关系/方体和主对比类关系/方体。
-
导出比较的表示
用可视化技术表达类比较描述,通常会包含“对比”度量,反映目标类与对比类间的比较。
3.实验过程
使用属性相关分析进行类特征化和类对比分析,使用Python语言完成编程实现。
(1)数据收集
使用给定的数据集进行类特征化和类对比分析,选定三类癌症BLCA,BRCA,KIRC进行分析。其中选定BLCA为目标类,BRCA,KIRC为对比类。这里直接使用原有的三个数据集,不做处理,待进行属性相关分析时再对数据进行需要的处理。
(2)预相关分析
这一步需要对有大量不同值的属性进行删除或概化,而且应尽量保守,保留更多属性用于后续的分析。对于癌症基因数据集,每个属性都是一个基因,要决定哪一个属性可以删除是困难的,一些属性或许可以根据概念分层向上攀升,但是需要对于基因信息具有专业知识才可以完成。例如:观察到一些基因id有相同的前缀,比如CXCL11|6373,CXCL14|9547,经查询资料,CXC是编码趋化因子蛋白质的基序,因此属性或许可以根据概念分层向上攀升(首先要将原数据的值进行一步概化,才能进行向上攀升的概化)。
考虑到数据集中的属性过多,难以删除或合并相等的概化的广义元组,这里不删除或以合并的方式概化任何属性。而是做最基本的概化,即将原数据集中各个基因的值概化为一个等级,划分原数据的范围区分不同等级,这类似于挖掘研究生的一般特征时,对GPA进行概化,根据GPA的分级作为概念分层。
对于数据集中数值的概化使用区间划分的方式进行概化,应该有一个标准,类似于成绩可以是低于60分划分为不及格,大于85分划分为优秀,这个标准应该是根据数据含义进行设置的。在癌症数据集中,这个值应该由了解数据的专业人员进行制定,这里仅为了练习与熟悉类特征化与类对比分析,因此简单的将数据进行等距划分,求出三个数据集中数据的最大值和最小值,划分为等距的区间。这一步比较简单,直接在EXCEL表格中完成,三个癌症类的数据集中最大的数据为4.62,最小为-1.85,划分为6个区间,每个区间长度为1.08。使用python程序完成概化,并保存到文件中。
核心代码如下:
step = 1.08
base_value = -1.86
base_path = "./属性概化结果"
# 对数据进行基本概化,将值概化为不同分级
def generalize(file_path:str):
file_name = os.path.basename(file_path) #文件名
output_path = os.path.join(base_path,file_name) #保存结果
df = pd.read_csv(file_path,index_col="gene_id")
df = pd.DataFrame(df.values.T,index=df.columns,columns=df.index) #进行转置
labels = list(df.columns.values) #gene_id
for index,row in df.iterrows():
for gene_id in labels:
for i in range(0,6):
if row[gene_id]>base_value+i*step and row[gene_id]<base_value+(i+1)*step:
row[gene_id] = i
break
df.to_csv(output_path)得到的结果形式如下:
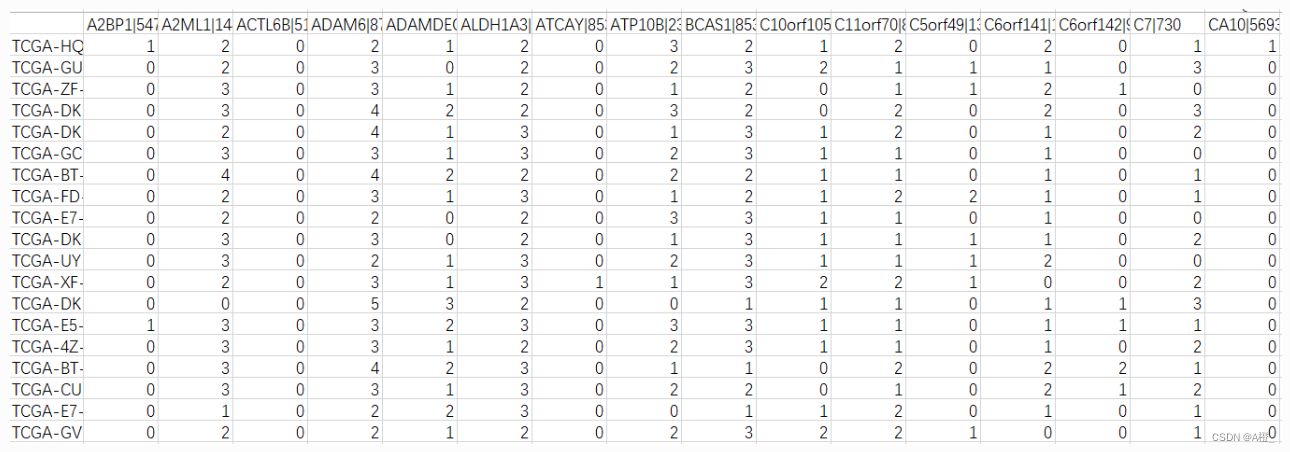
(3)基于信息增益的属性相关分析
完成了以上的属性概化后,计算每个属性的信息增益,去掉不相关或若相关的属性。
为了方便计算信息增益,先对概化后的数据进行统计,对每个基因不同的划分值的样本个数进行统计:
base_path = "./类特征化与对比分析数据"
def pre(file_path):
file_name = os.path.basename(file_path)
output_path = os.path.join(base_path,file_name)
df = pd.read_csv(file_path,index_col=0)
labels = list(df.columns.values)
data = {}
for gene_id in labels:
data[gene_id] = [0,0,0,0,0,0]
for index,row in df.iterrows():
for gene_id in labels:
data[gene_id][int(row[gene_id])] +=1
newdf = pd.DataFrame(data)
newdf.to_csv(output_path)
def main():
# 数据统计
pre("./属性概化数据/BLCArna.csv")
pre("./属性概化数据/BRCArna.csv")
pre("./属性概化数据/KIRCrna.csv")进行以上的统计后,统计数据形式如下:
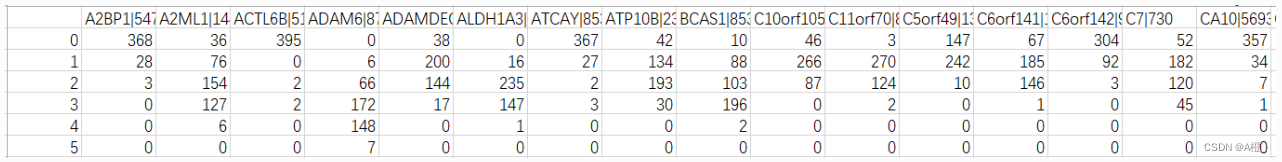
分类一个给定样本的期望信息计算如下:
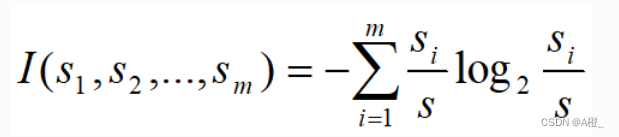
假设计算基因A进行分类的期望信息,一共有三个类,则s1,s2,s3为三个类在基因A上的样本数。计算如下:
# 计算分类一个给定样本的期望信息
def I(s:list)->float:
sum_s = 0
res_I = 0.0
for si in s:
sum_s += s
for si in s:
if si>0:
res_I += -(si/sum_s * math.log(si/sum_s,2))
return res_I如果样本根据基因A划分为v个子集,则计算给定的样本进行分类所需的期望信息(A的熵):
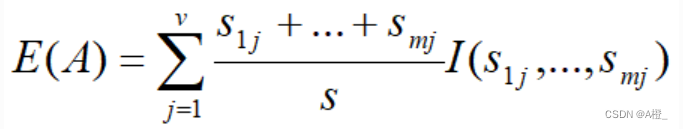
A上该划分的获得的信息增益为:
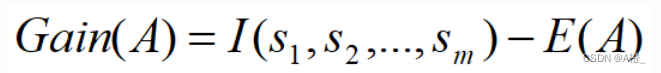
为方便数据处理,将E(A)的计算包含在Gain(A)的计算当中,完整的计算实现如下:
def main():
# 数据统计
pre("./属性概化数据/BLCArna.csv")
pre("./属性概化数据/BRCArna.csv")
pre("./属性概化数据/KIRCrna.csv")
df1 = pd.read_csv("./类特征化与对比分析数据/BLCArna.csv",index_col=0)
df2 = pd.read_csv("./类特征化与对比分析数据/BRCArna.csv",index_col=0)
df3 = pd.read_csv("./类特征化与对比分析数据/KIRCrna.csv",index_col=0)
result_path = "./类特征化与对比分析数据/Gain.csv"
# 计算每个属性的信息增益
labels = list(df1.columns.values) # 取出gene_id,接下来计算每个gene_id的信息增益
result = {}
for gene_id in labels:
gain = 0
# 先计算I(s1,s2,s3)
s = [0,0,0]
for i in range(6):
s[0] += df1[gene_id][i]
s[1] += df2[gene_id][i]
s[2] += df3[gene_id][i]
I_s = I(s) # 该基因整体对样本分类的期望信息
sum_s = s[0]+s[1]+s[2] # 该基因样本总数
# 计算E(gene_id)
E = 0
for i in range(6):
s1i = df1[gene_id][i]
s2i = df2[gene_id][i]
s3i = df3[gene_id][i]
silist = [s1i,s2i,s3i]
E += I(silist) * ((s1i+s2i+s3i)/sum_s) # 基因不同子集对样本分类的期望信息加权和即为属性的熵
gain = I_s - E # 该基因的信息增益
result[gene_id] = gain
resultdf = pd.DataFrame(result,index=["gain"])
# 转置,行为基因,列为信息增益
resultdf = pd.DataFrame(resultdf.values.T,index=resultdf.columns,columns=resultdf.index)
# 按信息增益从小到大排序
resultdf=resultdf.sort_values(by='gain',axis=0,ascending=True)
resultdf.to_csv(result_path)计算完成后的信息增益如下:
得到了各个属性的信息增益,就可以根据信息增益删除不相关和弱相关的属性了。对于不相关和弱相关的临界值的设定,如果属性总数较少,可以将这个临界值取小一些,保留较多的属性;如果像本数据集中,属性非常多,可能就需要将临界值取大一些,将大量弱相关的属性删除,尽可能保留强相关的属性。
绘制一个简单的密度图观察属性的信息增益情况:
df = pd.read_csv("./类特征化与对比分析数据./gain.csv",index_col=0)
df.plot.kde()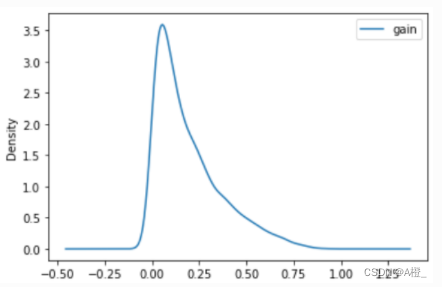
可以观察到绝大多数属性的信息增益都很小,小于0.25,因此实际上临界值取0.25就可以去掉大部分的弱相关和无关属性。由于该数据集本身的维度非常大,属性非常多,去掉弱相关和无关属性后仍有许多属性,在下面进行对比分析时,仅对强相关的属性进行比较分析,因此这里就不取具体的临界值进行属性删除了。
(4)特征化和对比分析
完成上述的属性概化和属性相关分析之后,就可以进行最后的特征化和对比分析了。可以采用量化规则,使用t_weight表示主概化关系中元组的典型性。
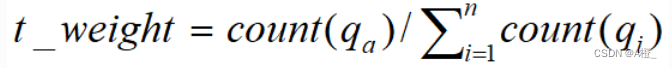
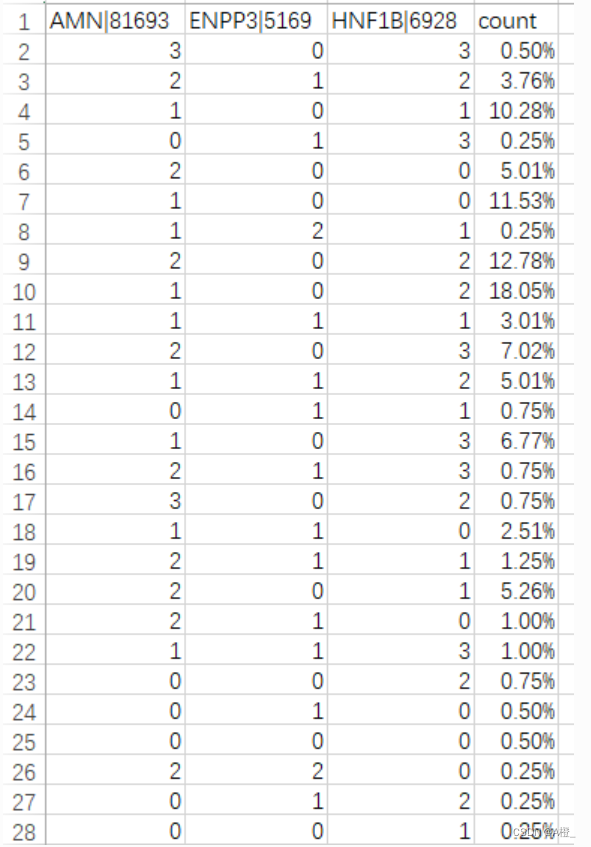
仅使用在属性相关分析中,信息增益最大的3个属性导出概化的表示和导出比较的表示。
result_path = "./类特征化与对比分析数据/characterize_discriminate.csv"
def main():
df = pd.read_csv("./类特征化与对比分析数据./gain.csv",index_col=0)
# 取信息增益最高的3个属性
gene_id = list(df.index[-3:-1])
gene_id.append(df.index[-1])
# 导出概化和比较的表示
data_count("./属性概化数据/BLCArna.csv",gene_id)
data_count("./属性概化数据/BRCArna.csv",gene_id)
data_count("./属性概化数据/KIRCrna.csv",gene_id)
def data_count(file_name,gene_id):
# 导出概化的表示
df = pd.read_csv(file_name,index_col=0)
# 原数据只保留指定的属性,然后进行count,这里只保留3个强相关属性
df = df[gene_id]
count = {}
total = len(df)
cols = gene_id.copy()
cols.append('count') # 保存count列
newdf = pd.DataFrame(columns=cols) # 结果
for row in df.iterrows():
item = tuple(row[1][gene_id]) # item即为这三个属性的一种取值组合
if item not in count:
count[item] = 1
else:
count[item] += 1
for key,value in count.items(): # 保存结果到文件
n_row = list(key)
n_row.append("%.2f%%" % (round(value/total,4)*100))
newdf.loc[len(newdf)] = n_row
newdf.to_csv(result_path,index = False, mode = 'a')导出结果如下,从上到下为BLCA,BRCA,KIRC的概化表示结果:
目标类BLCA的主概化结果:
类比较描述中的目标类和对比类的区分特性也可以用量化规则来表示,即量化区分规则:
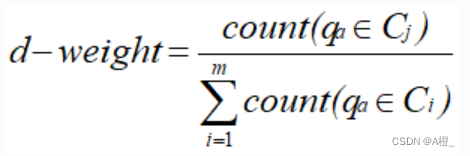
这和类特征化中计算t_weight的方式类似,只是计算初始目标类工作关系中的一种元组和目标类和对比类工作关系中这种元组的总元组数的比。实现如下:
def d_weight(gene_id):
# 计算d_weight,需要用到三个数据集中的数据
df1 = pd.read_csv("./属性概化数据/BLCArna.csv",index_col=0)
df2 = pd.read_csv("./属性概化数据/BRCArna.csv",index_col=0)
df3 = pd.read_csv("./属性概化数据/KIRCrna.csv",index_col=0)
# 保留指定属性
df1 = df1[gene_id]
df2 = df2[gene_id]
df3 = df3[gene_id]
count = {}
# 这里和特征化的过程是类似的,但是需要注意count的含义不同了,计算的是d_weight,此外结果还有一列为类型
cols = gene_id.copy()
cols.append('count') # count列
cols.insert(0,'type') # 增加一个类型列
newdf = pd.DataFrame(columns=cols)
# 计算d_weight,先各个类的概化元组数
for row in df1.iterrows():
item = tuple(row[1][gene_id])
if item not in count:
count[item] = [1,0,0] # 三个值表示目标类,两个对比类的该种元组数
else:
count[item][1] += 1
for row in df2.iterrows():
item = tuple(row[1][gene_id])
if item not in count:
count[item] = [0,1,0] # 对比类的概化元组
else:
count[item][1] += 1
for row in df3.iterrows():
item = tuple(row[1][gene_id])
if item not in count:
count[item] = [0,0,1]
else:
count[item][2] += 1
# 计算d_weight
for key,value in count.items(): # 保存结果到文件
n_row = list(key)
sum_num = value[0]+value[1]+value[2] # 总元组数
n_row1 = n_row.copy()
n_row2 = n_row.copy()
n_row3 = n_row.copy()
n_row1.append("%.2f%%" % (round(value[0]/sum_num,4)*100))
n_row2.append("%.2f%%" % (round(value[1]/sum_num,4)*100))
n_row3.append("%.2f%%" % (round(value[2]/sum_num,4)*100))
n_row1.insert(0,"BLCA")
n_row2.insert(0,"BRCA")
n_row3.insert(0,"KIRC")
newdf.loc[len(newdf)] = n_row1
newdf.loc[len(newdf)] = n_row2
newdf.loc[len(newdf)] = n_row3
newdf.to_csv("./类特征化与对比分析数据/d_weight.csv",index = False)结果形式如下:
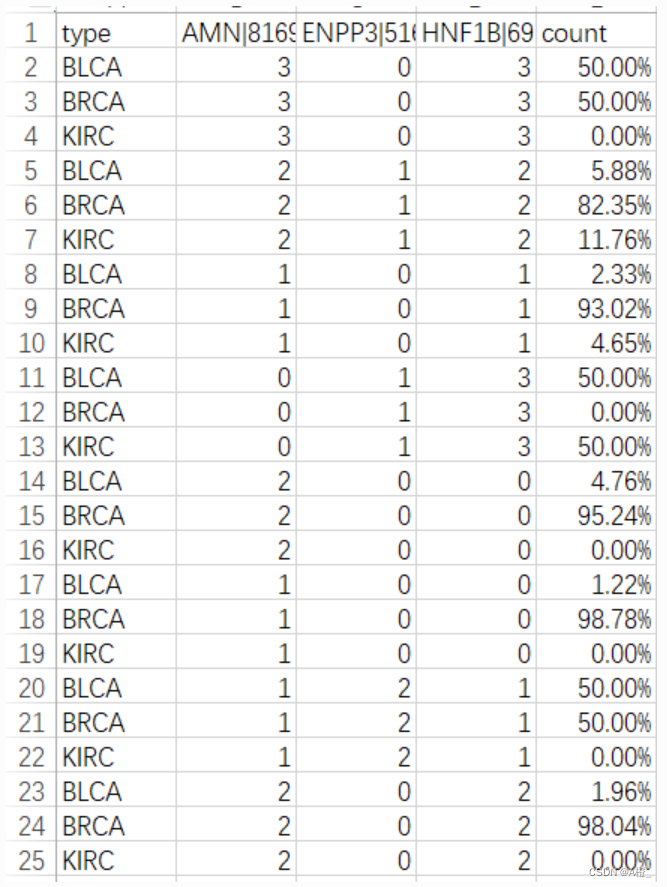
较高的count(即d_weight)值表示概化元组代表的概念主要来自于目标类。但最终的结果集中,目标类BLCA的d_weight值没有很高,反而是对比类BRCA和KIRC在很多概化元组上的d_weight接近100%,这可能意味着对比类的特征更加明显。
4.实验结果分析
对于特征化分析中的结果,目标类BLCA的特征似乎并不明显,概化元组的t_weight普遍比较低。而对比类BRCA和KIRC两类中,有一些t_weight值很高,例如BRCA在这三个属性上的等级为0,1,0(0对应原数据-1.85~-0.77,1对应原数据-0.77~0.31)的样本占了总样本的60%,可以认为是该类的重要特征。最后使用量化区分规则进行类对比分析时与类特征化时类似,目标类的d_weight不大,而对比类在一些概化元组上有显著的d_weight,这些概化元组可以明显的将目标类与对比类区分开。
处理过程中的一些简化对结果可能存在影响,例如属性概化时对值进行了区间划分,这个划分直接对最大值最小值之间进行等值划分,实际上可能刚好将体现特征的范围划分为了两部分,导致了结果不准确。
三.实验总结
通过本次实验,加深了对主成分分析,概念描述的类特征化和类对比分析的理解,并且进行了实现。第一题的主要问题为基因个数过多,远大于样本量,主成分分析的结果仍然很大,不确定如果样本充足,是否主成分分析的结果更好。主成分分析后对于结果该如何进行处理或分析还不了解。第二题主要是实现类特征化和类对比分析,最重要的部分是属性相关分析。在实现过程中主要是学习和尝试分析的方法和步骤,许多处理都简化且存在不合理,例如预处理进行属性概化时对值的区间划分,最后也只使用了根据计算信息增益得到的三个强相关的属性进行类特征化和类对比分析。尽管仍然存在许多问题(过程本身的问题,缺少对数据可视化展示的技巧等),但通过实验较为完整的完成了分析过程,理解了主成分分析,类特征化与类对比分析的目的和步骤。
智能推荐
2022-IOS-For-Fun_um-ios 2022-程序员宅基地
文章浏览阅读503次。2022 IOS Developer for funBasic stuffComputer Science fundamentalsMain parts of a computer system - CPU, memory, storageHow Operating System worksWhat is a databaseHow Internet worksGit version controlObject Oriented ProgrammingThe setupMacOSHomeb_um-ios 2022
PHP中的循环描述错误有哪些_PHP关于while循环中修改选取条件出现的错误-程序员宅基地
文章浏览阅读109次。业务需求是:读取某个表中每一行的的字段A、B、C的值如果C的值是0,就改成1或者2代码大概是这么写的:$query = "SELECT * FROM table WHERE C = 0";$result = mysqli_query($link, $query);if($result){while ($rows = mysqli_fetch_array($result)){if (判断条件为tru..._while循环报错php
ionic介绍-程序员宅基地
文章浏览阅读3.4k次。最近公司在使用ionic做混合APP,虽然是最后端,但是也查一下东西,介绍一下吧这是菜鸟教程的Ionic一.介绍ionic是一种老式的使用H5开发iOS和Android应用的方式,也可以使用新的语言React Native开发,当然对于H5实现复杂的或者交互性没有那么好的,就可以使用iOS和Android的插件实现;二.Ionic特点a.开发方面:1.ionic 基于Angular..._ionic
C/C++ 用什么软件编程_c++编程软件-程序员宅基地
文章浏览阅读3.7k次,点赞2次,收藏8次。Lightly 是一款全平台都通用、轻量且功能强大的在线编辑器,用户不需要配置任何编译运行环境,选择开发语言后即可开始写代码。代码和项目文件可以实时保存在云端的,换台不同系统的电脑或者是用平板,也可以打开项目,继续编码。它还具备项目一键分享功能,对于有团队协作需求的开发者来说,是最合适的选择。_c++编程软件
KEIL文件移动脚本--网关脚本_nrfutil' 不是内部或外部命令,也不是可运行的程序-程序员宅基地
文章浏览阅读650次。上面是效果原因 每次都是编译在大文件里面我希望只有BIN文件在一个干净的地方写一个.BAT在任何地方都是可以执行的 最后挂在KEIL里面内容 mv.batFOR /F %%I IN ('DIR /B /S "D:\TSBrowserDownloads\DA145xx_SDK_for_handover\DA145xx_SDK\old\projects\Izar\src\Node_Dialog_DA14531_SHENNONG\Keil_5\out_DA14531\Ob..._nrfutil' 不是内部或外部命令,也不是可运行的程序
全面解析并解决计算机缺失msvcp80.dll文件的问题-程序员宅基地
文章浏览阅读427次,点赞24次,收藏17次。在使用计算机过程中,有时会遇到“计算机缺失msvcp80.dll文件”的错误提示,这直接影响了部分应用程序的正常运行。msvcp80.dll是Microsoft Visual C++ 2005 redistributable runtime library(即VC++ 2005运行时库)的一部分,对于基于VC++ 2005编译的应用程序至关重要。本文将深入探究此问题产生的原因,并提出切实可行的解决方案。_msvcp80.dll
随便推点
Oracle 常用语句_oracle查询导入目录常用语句-程序员宅基地
文章浏览阅读112次。https://download.csdn.net/download/u014096024/21109113oracle练习1.如何查询一个角色包括的权限 a.一个角色包含的系统权限 select * from dba_sys_privs where grantee='DBA'; b.一个角色包含的对象权限2.oracle究竟有多少种角色 (查询oracle中所有的角色,一般是dba) select * from dba_roles;3.查询o..._oracle查询导入目录常用语句
数据可视化之美:经典案例与实践解析_数据可视化经典-程序员宅基地
文章浏览阅读9.3k次,点赞25次,收藏93次。随着DT时代的到来,传统的统计图表很难对复杂数据进行直观地展示。这几年数据可视化作为一个新研究领域也变得越来越火。成功的可视化,如果做得漂亮,虽表面简单却富含深意,可以让观测者一眼就能洞察事实并产生新的理解。可视化(visualization)和可视效果(visual)两个词是等价的,表示所有结构化的信息表现方式,包括图形、图表、示意图、地图、故事情节图以及不是很正式的结构化插图。基本的可视化展..._数据可视化经典
8086汇编4位bcd码_[走近FPGA]之二进制转BCD码-程序员宅基地
文章浏览阅读1.3k次。注:本文由不愿透露姓名的 @Bulingxx 撰写。以下为正文。在上一篇文章中介绍了数码管如何在FPGA开发板上实现动态显示,其文章链接如下:人生状态机:[走近FPGA]之数码管动态显示zhuanlan.zhihu.com本文的所有实例都使用硬木课堂Xilinx Aritx 7 FPGA板实现,且附有上板演示视频,该开发板的链接如下:硬木课堂 Xilinx Aritx 7 FPGA板 Arm C..._8086汇编语言 实现二进制数到bcd码的转换
使用nfs之后初始化mysql失败_influxdb数据库 nfs存储初始化失败-程序员宅基地
文章浏览阅读1.7k次。将nfs作为mysql的数据目录输出后,在另一台主机上启动mysql进程时,会出现如下这样的错误,究其原因,其实还是nfs自身设计的缺陷。 初始化就是使用特定的用户,去特定的目录去更新mysql,虽然说添加mysql用户之后,所有的对数据的修改权限都是以mysql用户执行的,而且nfs的数据目录也都设计成了mysql,常理是没有问题的。但是,执行mysql_ins_influxdb数据库 nfs存储初始化失败
ORC事务表与Hyperbase表的区别_星环 hyperbase、orc、text表区别-程序员宅基地
文章浏览阅读2.5k次。今天有客户问了我一下关于ORC事务表与Hyperbase表的区别问题,我回答的不是特别好,所以这里总结一下他们两个的区别,以便能掌握得更加深入些。ORC事务表:轻量级索引,支持CRUD操作,但是不建议大规模的单条增删改查,因为TDH(TDH是星环自研的一套大数据平台,类似于CDH,但是进行了很多的优化)是大数据数仓系统,是需要使用批量进行增删改查,索引单条操作的性能会降低;事务表需要进..._星环 hyperbase、orc、text表区别
Mybatis_"mybatis the content of element type \"choose\" mu-程序员宅基地
文章浏览阅读261次。Mybatis环境:JDK1.8Mysql5.7maven 3.6.1IDEA回顾JDBCMysqlJava基础MavenJunit1. 简介1.1 什么是MybatisMyBatis 是一款优秀的持久层框架它支持自定义 SQL、存储过程以及高级映射MyBatis 免除了几乎所有的 JDBC 代码以及设置参数和获取结果集MyBatis 可以通过简单的 XML 或注解来配置和映射原始类型、接口和 Java POJO(Plain Old Java _"mybatis the content of element type \"choose\" must match \"(when*,otherwise?)"